
Research Interests:
Our current research interests roughly fall into three distinct areas, protein folding, the influence of monomer structure and interactions on the thermodynamics of glass formation in polymer systems and thin, dense polymer films, and solvation and other complex phenomena in multicomponent self-associating systems. Our focus in these areas is on developing analytically tractable model that are useful in predicting new behavior, explaining perplexing data, and in guiding future experiments and the design of novel materials. We also pursue some simulations as necessary to test our theories and/or to guide the formulation of suitable approximations.
Many proteins in solution fold into beautiful structures that are intimately related to their function, while other proteins remain unfolded, presumably to enabling their functioning. We have developed a coarse-grained self-consistent model for predicting the folded structure and the mechanism of folding of small globular proteins without recourse, as is generally used in the field, to machine learning methods which are useful but which obscure the underlying physics. Our methods have been tested against the recent all-atom molecular dynamics simulation by D. E. Shaw and company for the dynamics of folding of twelve small, fast folding proteins using a supercomputer designed for MD simulations and built by Shaw, a hedge fund billionaire! Our simulations yield comparable results to Shaw (he gets better structures for six structures, while we beat him for the other six structures) but require on the order of 104 to 105 times less computer time (assuming that the MD simulations are performed on the same computer cluster as our coarse-grained simulations). Our current interests include protein folding in membranes, protein recognition, folding in larger proteins, and the nature of the poorly understood “structure” and behavior of intrinsically unfolded proteins.
The Nobel laureate P. W. Anderson stated in 1995 that the “deepest and most interesting unsolved problem in solid state theory is probably the theory of the nature of glass and the glass transition.” We have developed a theory for the influence of monomer molecular structure and interactions on the nature of glass-formation in glass forming systems, with particular emphasis on the glass transition Tg and the poorly understood fragility parameter m (a measure of temperature sensibility), the two material properties that are most important in dictating the method in which a material may be processed. Our predictions of the influence of the relative flexibilities of the backbone and side groups on the fragility of polymers with the structures of poly(n-α-olefins) have been verified by experiments as have been our predictions of the factors promoting anti-plasticization of glass forming polymers by the addition of small molecule diluents. Our current interests include devising methods for determine the parameters of our theory from experimental data, studying glass-formation in systems with strong, specific, self-assembling interactions, and more.
The development of a statistical mechanical theory for the thermodynamic properties of thin, dense polymers had remained an important unsolved problem in polymer physics, but we have recently devised a zeroth order lattice model theory of thin polymer films that is expected to be qualitatively correct but quantitatively crude. Our first goal is to devise methods for evaluating the higher order contributions in a systematic expansion for the properties of thin polymer systems. Subsequent work will study the influence of monomer molecular structure and interactions on the properties of polymer films.
The use of mixed solvents figures predominantly in methods for synthesizing and/or separate the components of a solution. Our interests focus on predicting the complex phase behavior that may emerge from the solvation of polymers in solutions of mixed self-assembling solvents. The possibilities are huge, and the resultant patterns can be highly non-intuitive, such as in the phenomena of cosolvency (cononsolvency), where a solute becomes solubility (insoluble) in a mixed solvent for which the solute is poorly (well) miscible in the individual pure solvents.
Recently, we have begun research on the influence of polarization in systems with charged, highly polarizing particles. Our theory provides insight into experiments displaying the aggregation dynamics of small micro-particles.
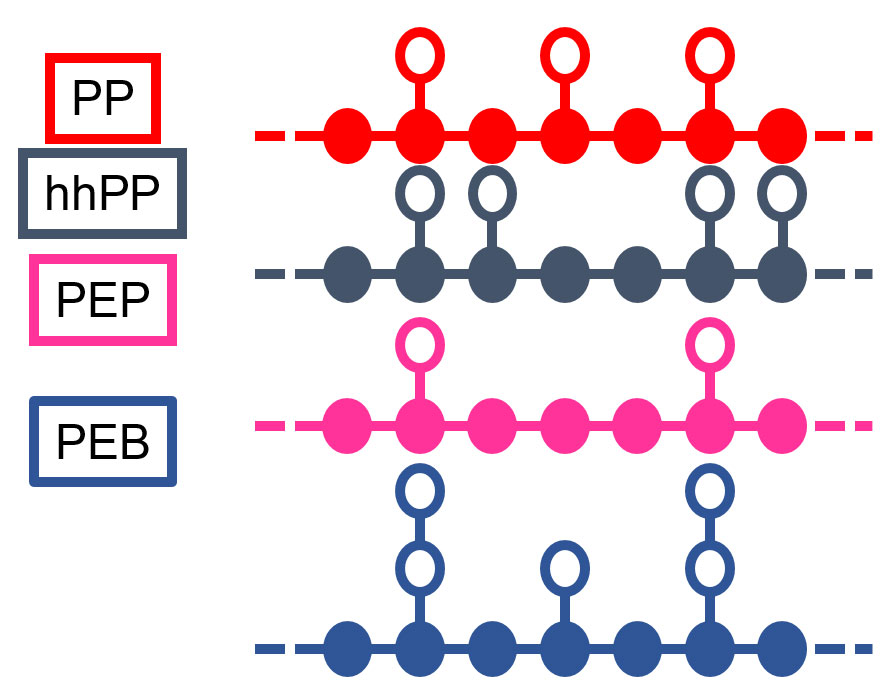
Fig. 1. Cartoon of monomer molecular structures used to study their influence on the miscibility and glass-formation in polymer systems.
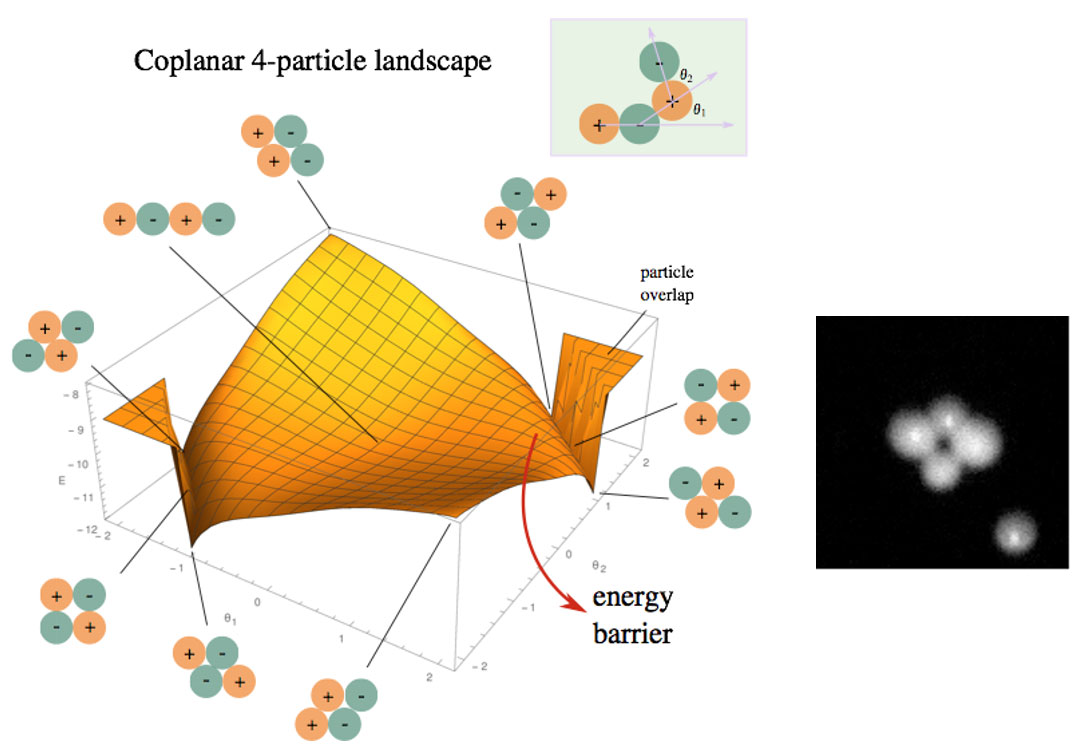
Fig. 2. Energy landscape including polarization (left) used to explain rearrangements of charged, polarizable four particle clusters (right) upon collision with fifth particle.
Selected References
Towards First Principles Theory of Relaxation in Supercooled Liquids Formulated in Terms of Cooperative Motion. K. F. Freed, J. Chem. Phys. 141, 141102 (2014).
Advances in the Generalized Entropy Theory of Glass-formation in Polymer Melts. J. Dudowicz, J. F, Douglas, and K. F. Freed, J. Chem. Phys. 141, 234903 (2014).
The Meaning of the "Universal" WLF Parameters of Glass-forming Polymer Liquids. J. Dudowicz, J. F, Douglas, and K. F. Freed, J. Chem. Phys. 142, 014905 (2015).
Lattice Cluster Theory for Dense, Thin Polymer Films. K. F. Freed, J. Chem. Phys. 142, 134901 (2015).
Theory of Competitive Solvation of Polymers by Two Solvents and Entropy-enthalpy Compensation in the Solvation Free Energy upon Dilution with the Second Solvent. J. Dudowicz, K. F. Freed, and J. F. Douglas, J. Chem. Phys. 142, 214906 (2015).
Even with Non-native Interactions, the Updated Folding Transition States of the Homologs Protein G and L are Extensive and Similar. M. C. Baxa, W. Yu, A. N. Adhikari, L. Ge, Z. Xia, R. Zhou, K. F. Freed, T. R. Sosnick, Proc. Natl. Acad. Sci. (U.S.), 112, 8302 (2015).
The Simplified Generalized Entropy Theory of Glass-formation in Polymer Melts. K. F. Freed, J. Chem. Phys. 143, 051102 (2015).
Cosolvency and Cononsolvency Explained in Terms of a Flory-Huggins Type Theory, J. Dudowicz, K. F. Freed, and J. F. Douglas, J. Chem. Phys. 143, 131101 (2015).
Entropy Theory of Polymer Glass-Formation in Variable Spatial Dimension. W.-S. Xu, J. F. Douglas, and K. F. Freed, Adv. Chem. Phys. 161, 443 (2016)
A Theory of Interactions between Polarizable Dielectric Spheres. J. Qin, J. Li, V. Lee, H. Jaeger, J. de Pablo, K. Freed, J. Colloid & Interface Sci. 469, 237 (2016).
Cooperative folding near the downhill limit determined with amino acid resolution by hydrogen exchange. W. Yu, M. C. Baxa, I. Gagnon, K. F. Freed, and T. R. Sosnick, Proc. Natl. Acad. Sci. 113, 4647 (2016).